Output
This describes the main output files of SNVPhyl. This includes the phylogenetic tree, SNV distance matrix, along with additional quality, filtering, and mapping information.
Phylogeny
The file phylogeneticTree.newick is the maximum likelihood phylogeny generated from an alignment of SNVs extracted from the whole genomes of each input file in Newick format.
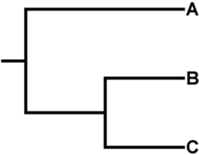
SNV/SNP Table
The file snvTable.tsv represents a table of all detected variant sites. The position of each site is given by a combination of Chromsome (contig/sequence name) and Position on the reference genome. The Status column represents whether this position was kept for constructing a phylogeny/distance matrix (valid) or filtered out. The possible status values are:
- valid: Represents a position that passed all filtering criteria for every genome. These positions are used in the SNV alignment used to construct a phylogenetic tree and a distance matrix.
- filtered-invalid: Represents a position that was removed due to either being present on a repeat region, within a region with a high-SNV density, or within a region passed in the
invalid_positionsfile. - filtered-coverage: Represents a position where at least one genome did not meet the minimum coverage criteria. The value inserted in the table in this case is a
-. - filtered-mpileup: Represents a position where there was a mismatch in variant calls between FreeBayes and SAMtools/mpileup/BCFtools. This often occurs in positions which met the min_coverage criteria, but did not meet the other criteria for calling a variant with FreeBayes, such as the minimum relative snv abundance (snv abundance ratio, or alternative allele proportion), or mapping quality scores. The value inserted in the table in this case is an
N.
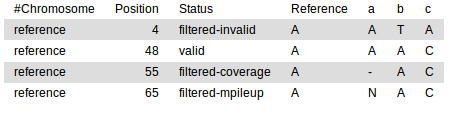
Filter Stats
A summary of the number of SNVs filtered within in the SNV Table.
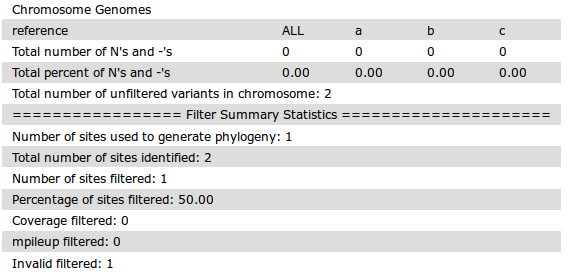
SNV/SNP Matrix
The file snvMatrix.tsv represents a pair-wise distance matrix of SNVs that passed all filtering criteria (has status valid in the snvTable.txt).
Core Positions
The file vcf2core.tsv is a table of the evaluated core positions in each reference fasta sequence.
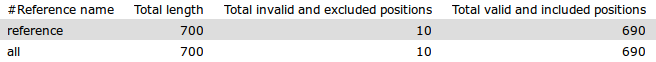
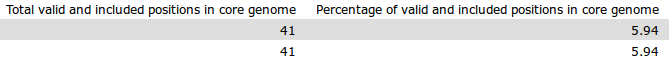
The columns are as follows:
- Reference name: The reference fasta sequence name. The value of all represents the sum of all sequences.
- Total length: The total length of the fasta sequence.
- Total invalid and excluded positions: The total number of invalid positions removed from analysis.
- Total valid and included positions: The total number of valid positions to be evaluated.
- Total valid and included positions in core genome: The total number of valid positions within the core genome. That is, the total positions with enough coverage to be evaluated.
- Percentage of valid and included positions in core genome: The percent of valid and included positions within the core genome. That is 100*(valid and included positions in core)/(valid and included positions).
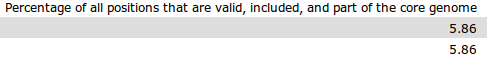
- Percentage of all positions that are valid, included, and part of the core genome: The percent of valid and included core positions out of the total length. That is 100*(valid and included positions in core)/(Total length).
SNV/SNP Alignment
The file snvAlignment.phy contains an alignment of SNVs used to generate the phylogenetic tree.

Mapping Quality
The file mappingQuality.txt describes how well the given reads mapped to the reference genome. This can help to evaluate if one genome has either too little data to be included in the analysis, or if the reference genome is very distantly related, or other potential issues.

The tool to generate this file takes two parameters, min coverage and min percent covered which represents the minimum percentage of the reference genome with reads over the minimum coverage mapped. The output is a list of all input genomes which did not meet the criteria as well as the percent of coverage over the reference genome.