Input
This describes the main input files of SNVPhyl. Please see the Usage documentation for details on running with these input files.
Reference Genome
The reference genome is used for the reference mapping stage of the pipeline. Ideally, this should represent a closed and finished genome very close to the other genomes to examine. When such as genome is not already available, it is possible to use a de novo assembled genome as a reference. However, with a de novo assembled genome there exists a greater possibilty of sequence alignment issues and missing data.
An example reference genome looks like.
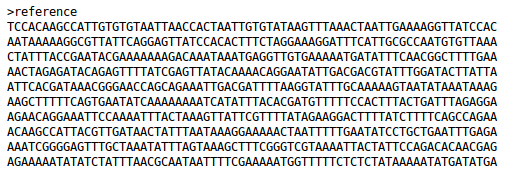
Invalid Positions masking file
The invalid positions file is used to mask out regions on the reference genome with variants. Masked regions will not be included in the phylogeny. The format of the invalid positions file is a BED-like tab-deliminated file defining the chromosome/contig/fasta sequence name and the start and end coordinates to exclude.
#Chromosome Start End
reference 1 10
The difference between this format and a true BED-formatted file is that coordinates are given starting with base 1 and are inclusive of the end position. That is, for this example, the positions defined start at the first base in a sequence (1) and include the end base (10).
Sequence Reads
The sequence reads are the set of reads to use for mapping and variant calling. The provided workflows support both paired-end and single-end sequence reads but not a mixture of both in the same workflow. These should be in FASTQ format. Within Galaxy these should be imported with the fastqsanger data type and structured within a dataset collection of type list or list of paired-end files.
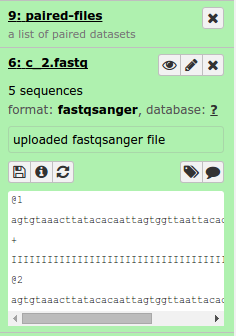
Please see the Preparing Sequence Reads documentation for details on how to structure the data.