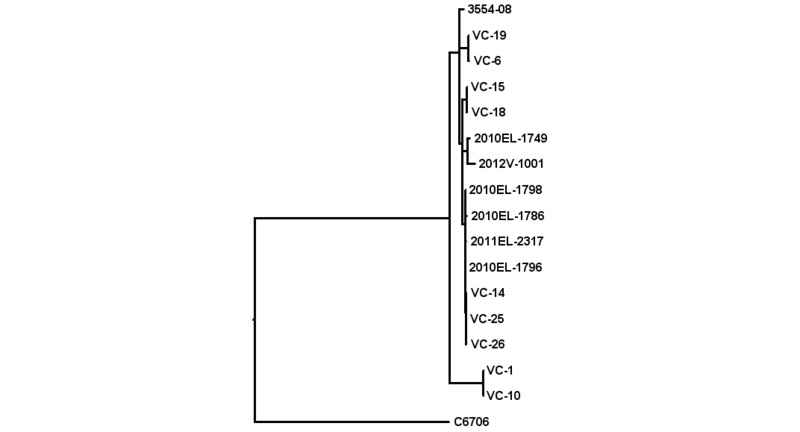
SNVPhyl: Whole Genome SNV Phylogenomics Pipeline
The SNVPhyl (Single Nucleotide Variant PHYLogenomics) pipeline is a pipeline for identifying Single Nucleotide Variants (SNV) within a collection of microbial genomes and constructing a phylogenetic tree. Input is provided in the form of a collection of whole genome sequence reads as well as an assembled reference genome. The output for the pipeline consists of a whole genome phylogenetic tree constructed from the detected SNVs, as well as a list of all detected SNVs and other information.
Operation
SNVPhyl identifies variants and generates a phylogenetic tree by mapping the input sequence reads to a reference genome followed by filtering out any invalid variant calls. The stages are as follows:
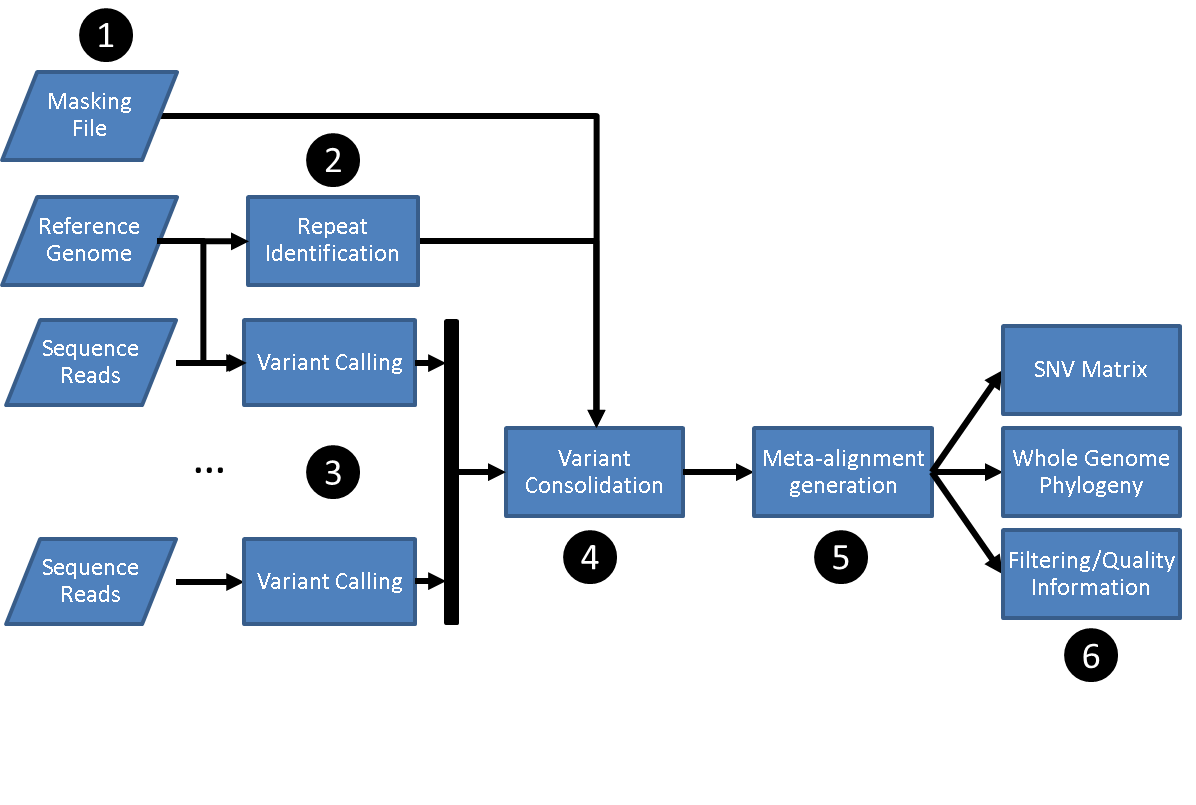
- Preparing input files including:
- A set of sequence reads.
- A reference genome.
- An optional file of regions to mask on the reference genome.
- Identification of repeat regions on the reference genome using MUMMer.
- Reference mapping and variant calling using SMALT, FreeBayes and SAMtools/BCFtools.
- Merging and filtering variant calls to produce a set of high quality SNVs.
- Generating an alignment of SNVs.
- Building a maximum likelihood tree with PhyML and generating other output files.
SNVPhyl is implemented as a Galaxy workflow, with each of these stages implemented using a specific Galaxy tool.
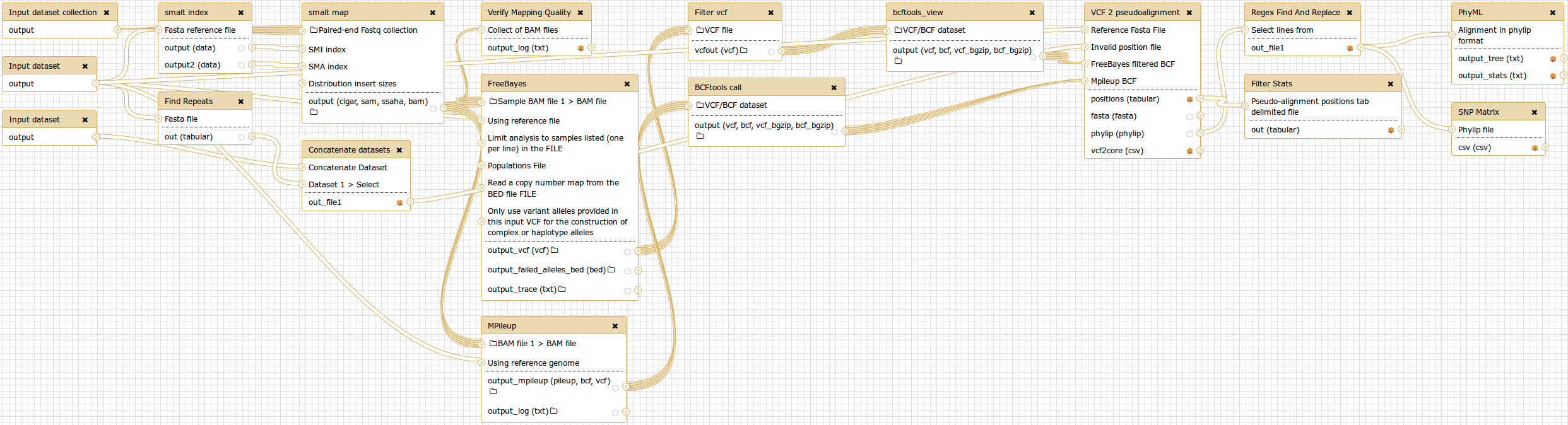
More information on the operation and installation of the pipeline can be found in the Usage and Installation sections.
Contact
Comments, questions, or issues can be sent to Aaron Petkau - aaron.petkau@phac-aspc.gc.ca.