SNVPhyl: Whole Genome SNV Phylogenomics Pipeline
The SNVPhyl (Single Nucleotide Variant PHYLogenomics) pipeline is a pipeline for identifying Single Nucleotide Variants (SNV) within a collection of microbial genomes and constructing a phylogenetic tree. Input is provided in the form of a collection of whole genome sequence reads as well as an assembled reference genome. The output for the pipeline consists of a whole genome phylogenetic tree constructed from the detected SNVs, as well as a list of all detected SNVs and other information. The pipeline is implemented using the Galaxy bioinformatics analysis platform.
Quick Usage
To quickly get started with SNVPhyl, a command-line interface is provided to interact with Galaxy. This can be configured to deploy a Docker image which includes all the SNVPhyl tools and an instance of Galaxy.
git clone -b master https://github.com/phac-nml/snvphyl-galaxy-cli.git
cd snvphyl-galaxy && pip install -r requirements.txt
python bin/snvphyl.py --deploy-docker --fastq-dir example-data/fastqs --reference-file example-data/reference.fasta --min-coverage 5 --output-dir output1
This assumes that both Python and Docker are installed. If Docker requires sudo to run, please append --with-docker-sudo to the above command.
Alternatively, the Docker instance can be launched directly with:
docker run -d -p 48888:80 phacnml/snvphyl-galaxy-1.0.1
Once running, SNVPhyl/Galaxy can be accessed from http://localhost:48888 with username admin@galaxy.org and password admin. Please see Usage and Installation for more details.
Nextflow
A version of SNVPhyl developed for Nextflow by Jill Hagey is available at https://github.com/DHQP/SNVPhyl_Nextflow.
Operation
SNVPhyl identifies variants and generates a phylogenetic tree by mapping the input sequence reads to a reference genome followed by filtering out any invalid variant calls. The stages are as follows:
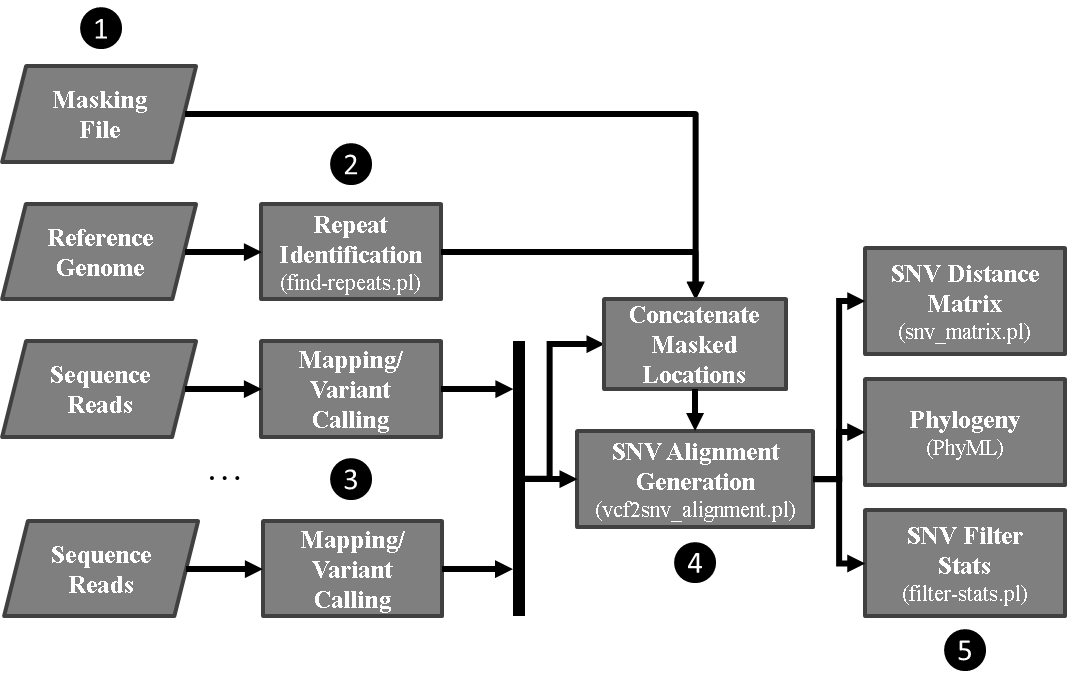
- Preparing input files including:
- A set of sequence reads (
fastqformat). - A reference genome (
fastaformat). - An optional file of regions to mask on the reference genome (a special tsv formatted file).
- A set of sequence reads (
- Identification of repeat regions on the reference genome using MUMMer.
- Reference mapping and variant calling to identify high-quality SNVs (hqSNVs) using SMALT, FreeBayes and SAMtools/BCFtools.
- Merging the identified hqSNVs to construct a multiple sequence alignment.
- Building a maximum likelihood tree with PhyML and generating other output files.
SNVPhyl is implemented as a Galaxy workflow, with each of these stages implemented using a specific Galaxy tool.
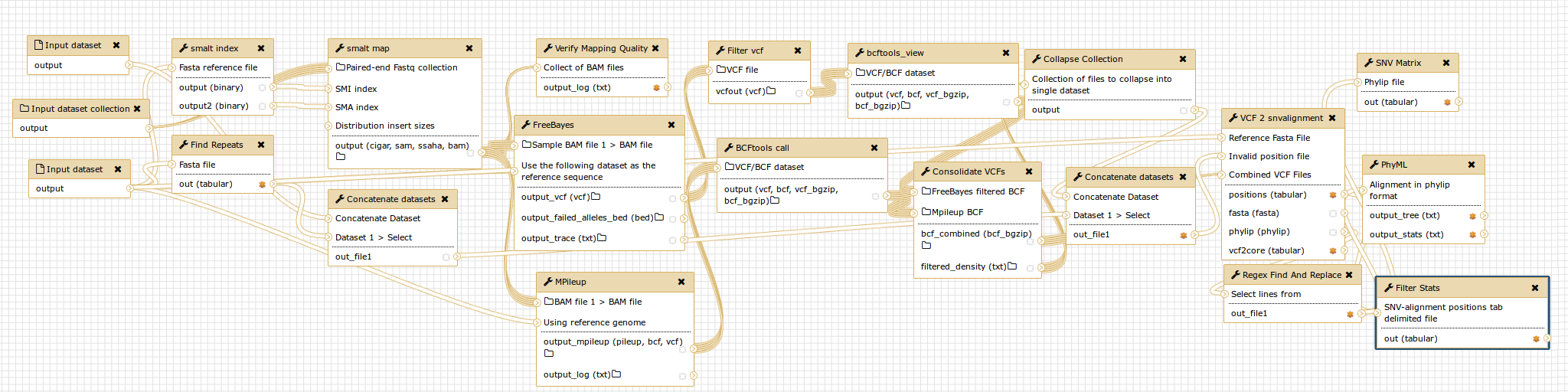
More information on the operation and installation of the pipeline can be found in the Usage and Installation sections.
Code is available on GitHub under the https://github.com/phac-nml/snvphyl-galaxy, https://github.com/phac-nml/snvphyl-tools, and https://github.com/phac-nml/snvphyl-galaxy-cli projects.